







Next Generation Sequencing and National Health Service
What data should we retain after variant reporting?
This is a Press Release edited by StorageNewsletter.com on February 1, 2016 at 2:57 pmHere is an article by Kevin Blighe, Ph.D., and Darren Grafham, Sheffield Children’s National Health Service (NHS) Foundation Trust, Western Bank, Sheffield, UK.
Next generation sequencing and the National Health Service:
what data should we retain after variant reporting?
Abstract
This report discusses issues of data storage for next generation sequencing (NGS) within the clinical diagnostic environment of the National Health Service (NHS). We describe disk space requirements for a sequence run and per sample, and the long term data accumulation of these. We also consider the cost-benefit of storing all data versus storing data from various stages in the analysis process and regenerating results, including sequencing samples afresh. The impact of changing technologies and improvements in analytical methods are discussed, along with the ease of retrieving data and updating analyses as additional knowledge of disease pathways is gained.
Background and introduction
Next generation sequencing technology has become an important tool in research and is now being implemented in clinical diagnostic environments where it is hoped to bring improved cost- and time-savings. Indeed, NGS has great potential to be translated into improved patient care. Validated equipment and processes (or workflows) are the cornerstone of the use NGS in such settings. For example, sequencer instruments that are used must have prior approval for use as diagnostic tools. In Europe, this comes in the form of regulatory approval through the European Medicines Agency (EMEA), whilst, in the United States of America (USA), it is the Food and Drug Administration (FDA) that makes such approvals. In addition, before the implementation of a ‘live’ NGS service, there must be a consensus on which analytical pipeline to use to process the data and produce variants, with follow-up confirmation of these with the current gold standard in clinical diagnostics, i.e. Sanger sequencing. Such analytical pipelines must also adhere to data protection and patient privacy standards, like BS7799 in the United Kingdom of Great Britain and Northern Ireland (United Kingdom), and also variant reporting standards set by the Human Genome Variation Society (HGVS).
The fundamental data-type that is created during a NGS workflow is comprised of the raw instrument data, i.e., the image files and signal intensities. If these files were to be stored for long periods of time, disk storage mediums with greater capacities would be required on sequencer instruments and, thus, greater cost (including maintenance cost). Currently, this is overcome through the shipment of high disk capacity servers with such sequencers (i.e. these servers are included in the fixed cost of the sequencer itself). As the sequencing process proceeds, the raw data that is generated is automatically transferred onto the attached server; however, these servers still contain an unfeasible amount of storage capacity when even 1+ years worth of sequencing on a regular basis – like in a clinical diagnostic laboratory – is a consideration. In addition, this is only taking into consideration targeted sequencing and not exome or whole genome sequencing. In such scenarios, this raw instrument data may not even be stored at all.
Even in the realm of targeted sequencing, if a laboratory manages to implement a validated NGS workflow, its use on a continuous basis – like it would be in a clinical diagnostics department – can result in an increased strain on disk storage. For example, at our department in the Sheffield Children’s NHS Foundation Trust, where NGS is currently being used as a live service, the service has generated a total of 6.2TB of data since its inception in the second quarter of 2013[1]. This is divided between results files (1.2TB), data generated prior to the NGS service going live (2.3TB), raw instrument data and base-calls (2.7TB), and files required to run the analytical pipeline (0.021TB) (Figure 1). With such a large accumulation of data in such a short time-frame, laboratories will be forced to think about how they can best cope with disk storage requirements in the short- and long-term future when using NGS as part of a live diagnostic service. The long-term integrity and ongoing accessibility of this stored data is also critical, a requirement coined as ‘digital preservation’ by Arkivum Ltd[2]. Failure to include a digital preservation strategy from the outset of a NGS service can compromise the integrity of the data and result in loss of clinical information. Whilst saying this, as NGS technology is still relatively new, it is recognised that it has been difficult to predict the exact requirements that are needed for using NGS, either in a research or clinical setting.
Figure 1 – Overview of disk usage in our NGS service
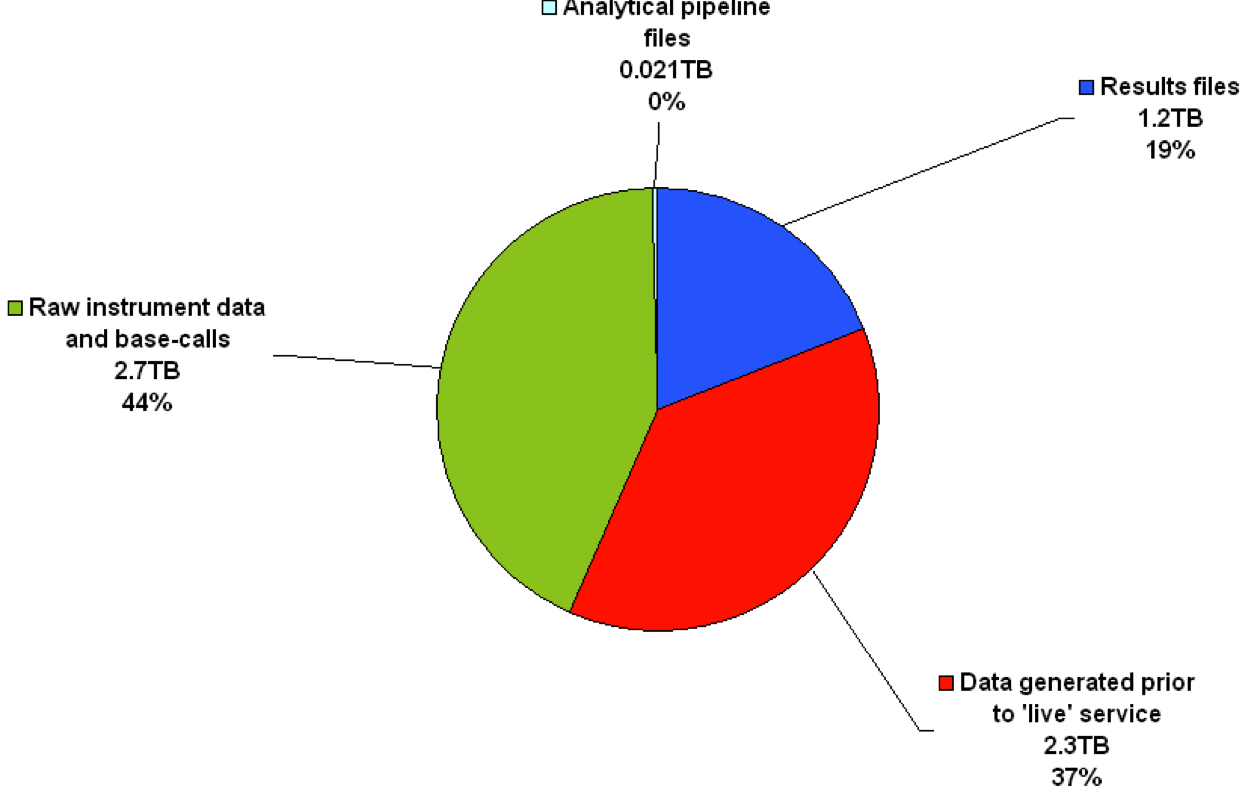
The pie chart shows the distribution of disk usage per data-type for our department’s NGS service. The raw instrument data and base-calls combined comprise the majority (2.7TB, 44%), whilst results files (i.e. some of the files generated by the analytical pipeline and also the base-calls in this case) comprise less than half of this (1.2TB, 19%). In total, 6.2TB has been stored on our servers since NGS was introduced into the department (roughly 18 months).
Sequencers in NGS are also capable of running multiple samples in the same reaction – called ‘barcoding’ – which reduces costs on reagents by a great deal. Typically, 16 or 32 samples can be pooled in the same reaction, but this number can be increased multiple times higher. Thus, the NGS workflow is a highly streamlined process: through the barcoding process, NGS sequencers can be used more frequently and for cheaper costs. The main bottlenecks in the workflow are still with the analysis and interpretation of the data, let alone dealing with the storage requirements. This situation – i.e., where sequencers are highly streamlined but the methods post-sequencing to process all of the data are somewhat less so – has been coined ‘analysis paralysis’ (doi:10.1038/nmeth0710-495).
Costs of disk storage versus DNA sequencing
Up until the introduction of NGS, the long-term trend in cost of sequencing a single base-pair (1bp) remained higher than that of storing 1MB of information. With the introduction of NGS, however, the cost of sequencing 1bp dropped sharply and eventually become cheaper than the storage cost of 1MB (Figure 2). Thus, although NGS has made DNA sequencing cheaper, disk storage costs for NGS data – a type of data that requires much greater disk capacities than traditional methods of sequencing – have not dropped at the same rate, leaving such storage costs relatively high.
Figure 2 – Inverse in trends over time in the costs of disk storage versus DNA sequencing
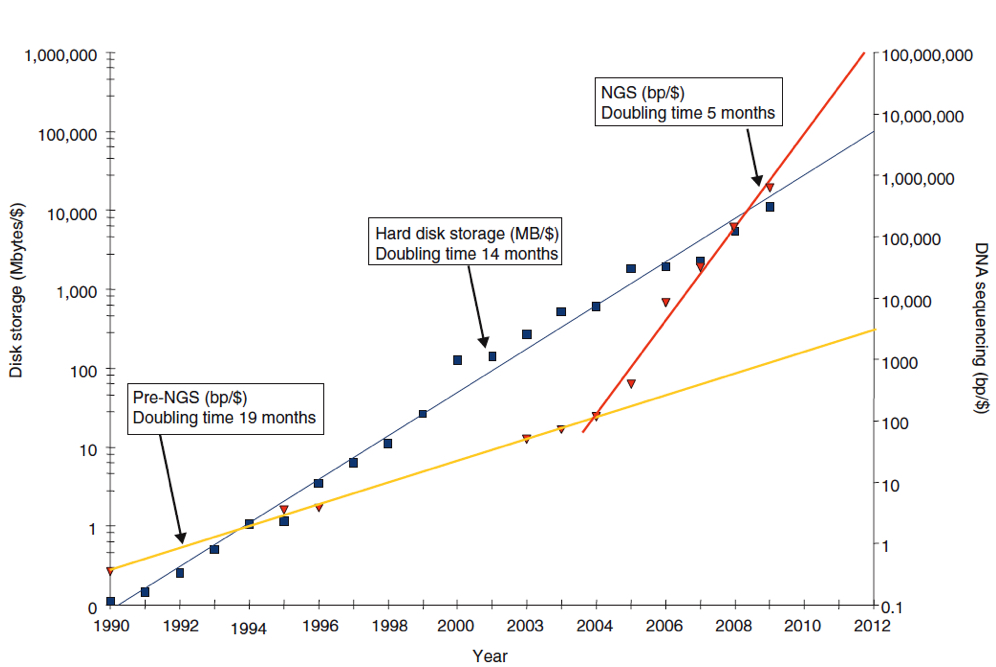
The line graph, plotted as inverse, shows an exponential reduction over time for both disk storage and DNA sequencing, with disk storage costs falling slightly quicker than those for DNA sequencing. However, the introduction of NGS (~2004) resulted in a sharper decrease in DNA sequencing costs, gradually surpassing those of disk storage (i.e. become cheaper) in around 2009, leaving disk storage costs relatively high. Blue squares, cost of storing 11MB per US dollar; blue line, exponent of cost of storing 1MB/$; red triangles, cost of sequencing 1 base-pair (1bp)/$; yellow line, exponent of DNA sequencing costs (in bp/$) prior to introduction of NGS; red line, exponent of DNA sequencing costs (in bp/$) after the introduction of NGS (doi:10.1186/gb-2010-11-5-207).
Data-types in NGS
Data produced from NGS technology and an analytical pipeline include (see also Figure 3 and Table 1):
- Raw instrument data (image files and signal intensities)
- Sequencing-associated files (log files, quality metrics, setup information)
- Base-calls (FASTQ, FASTA, BAM, etc.)
- Alignments (BAM, SAM, etc.)
- Filtered alignments (BAM, SAM, etc.)
- Quality metrics (percent alignment, statistics on coverage, percent reads on target, etc.)
- Variant calls (VCF)
- Filtered variant calls (VCF)
- Customised report for distribution (TXT, DOC, PDF, etc.)
- Analytical pipeline record (details of analysis sufficient for an exact reanalysis, including software versions, database versions, etc.)
Figure 3 – Overview of data generated by a NGS sequencer and an analytical pipeline
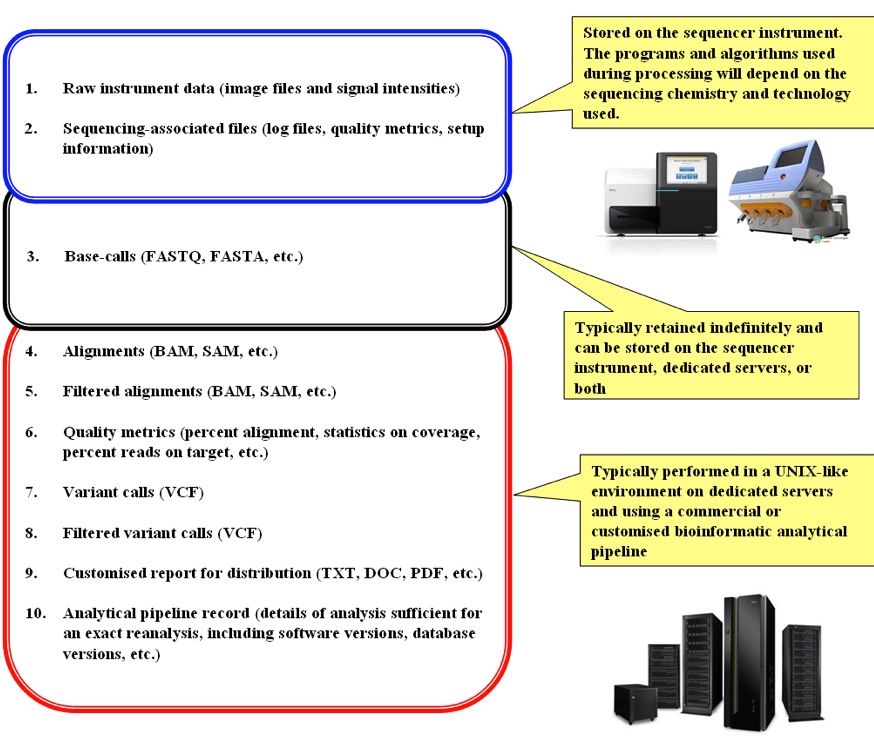
The figure shows the general flow of events from the production of the raw instrument data to the generation of results in the final report for distribution, along with the different types of data produced by each step and notes on their typical storage locations. Extra information on each data-type is found in Table 1.
Table 1 – Overview of data generated by a NGS sequencer and an analytical pipeline
The table provides general information on each type of data produced during the different stages of a NGS analytical pipeline (as per Figure 3), including a description of the data format in each case and a rough guide to the size on disk across 3 main NGS sequencer instruments.
|
Data-type |
Description of data-type |
Size on disk |
||
|
MiSeq™ |
HiSeq™ |
Ion Torrent™ |
||
|
Raw instrument data (image files and signal intensities) |
The image files are graphical files, whereas the signal intensities are data-matrices that attempt to encode these images numerically. |
|||
|
Sequencing-associated files (log files, quality metrics, setup information) |
ASCII files giving information for each run on the sequencer. |
|||
|
Base-calls (FASTQ, FASTA, etc.) |
Base-call files are generated from the signal intensities and contain the base-calls inferred from this data. The base-calls are assembled into separate reads, with each read having an identifier. The FASTQ and FASTA formats are both ASCII but are typically compressed or zipped. They both store the read identifier and the called bases pertaining to each; however, FASTQ additionally stores quality information (as ASCII) for each base-call and is unfiltered. Generally, FASTQ data is filtered on base quality to produce FASTA, which may therefore have reads and/or bases removed. FASTQ: Line 1, read identifier; Line 2, base-calls; Line 3, optional identifier; Line 4, quality of each base in Line 1 @M00969:109:000000000-AAD9T:1:1101:15890:1648 1:N:0:4 !”*(……(((***+) FASTA: Line 1, read identifier; Line 2, base-calls >M00969:109:000000000-AAD9T:1:1101:15890:1648 1:N:0:4 AGGTA……GAAACTGG |
|||
|
Alignments (BAM, SAM, etc.) |
The SAM format is the sequence alignment map format and contains information on each read after having conducted an alignment to a reference genome. These files are ASCII and are comprehensive in what they encode. BAM is the binary equivalent of a SAM file, generally used to save disk space. |
|||
|
Filtered alignments (BAM, SAM, etc.) |
Using information contained in a SAM/BAM file, reads can be filtered out or marked for exclusion whilst preserving the SAM/BAM specification. |
|||
|
Quality metrics (percent alignment, statistics on coverage, percent reads on target, etc.) |
Typically ASCII files giving information for the data contained in the SAM/BAM files. |
|||
|
Variant calls (VCF) |
The VCF format is the variant caller format and contains a list of variants against the reference used during the alignment step for a particular sample or samples. For each variant, additional information can also be specified, such as annotation information, zygosity, depth, quality of the variant call, etc. These files are ASCII but can be compressed / zipped. |
|||
|
Filtered variant calls (VCF) |
Using information contained in a VCF file, variants can be filtered out whilst preserving the VCF specification. |
|||
|
Customised report for distribution (TXT, DOC, PDF, etc.) |
Using all information gathered in the analytical pipeline, a customised report can be generated in any chosen format. |
|||
|
Analytical pipeline record (details of analysis sufficient for an exact reanalysis, including software versions, database versions, etc.) |
Typically an ASCII file(s) that contains enough information such that the analytical pipeline couple be re-initiated to produce the same results each time. |
|||
Current recommendations
There is currently no consensus or standard for storing data produced by NGS technology. Adherence to data protection and patient privacy laws is essential; there is no specific legal framework within which to operate. In recognising that different clinical diagnostic laboratories may employ different analytical pipelines – even when using the same sequencer instrument – the American College of Medical Genetics (ACMG) state that such laboratories ought to themselves specify the file types to be stored and, moreover, the length of time that each is to be stored (doi:10.1038/gim.2013.92). In the United States of America, there is some legal framework for storing ‘analytic systems records’ and ‘test reports’ for at least 2 years (CLIA regulations, section 493.1105). In the context of NGS, the ACMG adopt this framework and recommend the storing of data that would “allow regeneration of the primary results as well as reanalysis with improved analytic pipelines” for at least 2 years. As virtually all analytical pipelines in NGS begin with the base-call files, this would entail the storing of the FASTQ and FASTA files and would allow a replicate analysis using the same pipeline, as well as comparison with an improved analytical pipeline. The ACMG also recommend the additional storage of the results file (VCF) for “as long as possible“, citing the future likelihood of a request for reinterpretation of the results as being significant in making this recommendation. However, solely storing these data-types would not allow for base-calling using a different base-calling algorithm to be performed. Such algorithms can differ in the statistical methodologies used and the results produced (doi:10.1093/bib/bbq077; doi:10.1093/bioinformatics/btt117); moreover, a software update on a sequencer instrument that incorporated a change in the base-calling algorithm could be critically important. As such, the case can be made that the fundamental data-type that should be stored is actually the raw instrument data, but – as mentioned – the files that comprise this data are large. This can be seen in our laboratory, where a total of 1.2TB of results files have been generated since the inception of our NGS service, yet a total of 2.7TB of raw instrument data and base-calls have been generated concurrently.
Thus, adhering to the ACMG recommendations, it is only the base-calls and the results files that ought to be stored. This excludes all other types of data produced by NGS technology, i.e., the raw instrument data, alignments, intermediary files, annotated results, and customised reports for distribution.
Other recommendations pertaining to the storage of NGS data have been made. A noteworthy mention must go to Arkivum Ltd. who compiled a list of 10 recommendations for the long-term storage of NGS data, with particular focus on large-scale projects like the upcoming Genomics England Ltd. (GEL) sequencing of 100,000 whole human genomes. Their recommendations are summarised (Figure 4). Indeed, such large-scale and publicised projects require the utmost of planning prior to their commencement. Although not entirely relevant to sequencing in the realm of clinical diagnostics, the recommendations are prudent and comprise a helpful guide on ensuring that the long-term integrity and ongoing accessibility of data is made. This includes adherence to the open archival information system (OAIS) standard (ISO 14721:2012) and the use of a trusted digital repository (TDR)[3].
[1] An organisation can achieve TDR status after having gained the Data Seal of Approval (DSA) by the Data Archiving and Networked Services (DANS).
Figure 4 – Ten recommendations for the long-term storage of NGS data

Recommendations are made with a particular focus on large-scale projects, such as the sequencing of 100,000 whole human genomes by GEL. Adapted from: Arkivum Ltd., Executive Summary: GEL Data and Metadata Preservation (Version 1.0A), August 2014.
Although useful to follow in order to ensure the long-term integrity and ongoing accessibility of NGS data, these recommendations fall short of saying where the most ideal location to store data should be. Particularly in a clinical setting, deciding on a storage location is of high importance.
So, where to store the data?
The easiest way to ensure the rapid transfer and security of data is to store it locally, i.e., ‘in house’ and within the confines of the network within which they were created. Such a network should have in-built redundancy, such as RAID, and ideally be monitored by trained IT personnel, such as a network administrator. Indeed, the key involvement of IT personnel is a must in this regard.
The ACMG mention the use of cloud computing services as alternatives, but at the same time cast doubt on their use. As such services are not compliant with the Health Insurance Portability and Accountability Act, they do not allow for the traceability of patient data. In addition, it may not prove utile to upload and download such large amounts of data from a remote cloud server unless a large amount of bandwidth is at your disposal. On top of this, these services are usually charged.
Conclusive recommendations from this report
Taking everything into consideration, it is of paramount importance that the raw instrument data is retained indefinitely, particularly in a clinical setting. The algorithms used to process this raw instrument data and convert it into base-calls are platform-dependent, i.e., they differ depending on the sequencing technology employed, and they can be modified. Indeed, vendors of sequencer instruments periodically update these algorithms; thus, the possibility exists that a ‘better’ or ‘improved’ algorithm was introduced that could alter the results for a sample being sequenced. Although retaining just the base-calls would allow for the majority of a NGS workflow to be repeated, it would still skip this initial step (raw instrument data conversion into base-calls) where, potentially, a base could be called differently or at a different quality. In the absence of the retention of such raw instrument data, a sample aliquot could alternatively be repeated in order to employ updated algorithms, but this is assuming that an extra aliquot is available, which may not be the case. Repeating a sample in this way could also introduce bias from the wet laboratory.
To conclude, we suggest the following:
- Archive the raw instrument data for each run indefinitely
- Locally store, for each sample, all other data as defined in a MIMIM for at least 2 years, including:
- Quality control reports
- Results file (unannotated and unfiltered)
- Customised report
- Log file that contains sufficient information to allow for an exact replicate of the results to be reproduced from the raw instrument data (i.e. detailing all commands and program versions used in the analytical pipeline)
- Plan for the digital preservation of all stored data and involve either: 1, a third-party TDR; or 2, the local IT department and/or a designated network administrator
Competing interests
We declare the following interests: None
[1] Next generation sequencing was introduced into the department in the second quarter of 2013 but went ‘live’ in the final quarter of the same year.
[2] Arkivum Ltd., Executive Summary: GEL Data and Metadata Preservation (Version 1.0A), August 2014.
[3] An organisation can achieve TDR status after having gained the Data Seal of Approval (DSA) by the Data Archiving and Networked Services (DANS).




 Subscribe to our free daily newsletter
Subscribe to our free daily newsletter
